Details To be Announced
Project funding: University of Groningen, NL
[Photo credit: xxx]
CURRENT TEAM MEMBERS

Bioinformatician
MIVEGEC, U. Montpellier, FR
PhD student (coll V Rougeron & F Prugnolle)
"Popualtion genomics and invasion history Plasmodium parasites".
MIVEGEC, U. Montpellier, FR

PhD Student
U. Groningen, NL
PAST TEAM MEMBERS (PhD and Postdocs)
Former PhD student with me at GELIFES, U. Groningen, NL
Now post-doc/bioinformatician @ U. Harvard (Prof. DE Neafsey)

Former PhD student with me MIVEGEC (Coll. A-S Fiston Lavier, ISEM)
MIVEGEC, U. Montpellier, FR

Former PhD student with me at GELIFES, U. Groningen, NL
Departing for the U. Sheffield, UK

Former PhD student with BEK Eriksson, ME Maan at GELIFES, U. Groningen, NL

Former PhD student with me and J Flacao-Salles and L Beukeboom @GELIFES (U. Groningen)
Now postdoc @German Centre for Integrative Biodiversity Research (iDiv) Halle-Jena-Leipzig | iDiv

Former Post doc researcher with me and NJ Besansky (U Notre Dame, IN, USA)
Currently Postdoc at the U. Chicago, USA

Former post-doc researcher with JL Olsen and me
Currently at ...

Former post-doc researcher with O Gaggiottii, AE Foote, and me
Currently postdoc at the U. Copenhagen, DK
PAST TEAM MEMBERS

Former Intern Student
Now PhD student @ MIVEGEC
CURRENT PROJECTS
Porpoise ecological and evolutionary genomics

PI : Michael C. Fontaine
Collaborators: Phil Morin (SWFC, NOAA, CA USA), Yacine Ben Chehida (U. Gronginen, NL)
learn more ...
Collaborators: Phil Morin (SWFC, NOAA, CA USA), Yacine Ben Chehida (U. Gronginen, NL)
learn more ...
Genomic perspective of the parallel evolution to coastal habitat in the bottlenose dolphins

PIs : Oscar Gaggiotti (U. St Andrews, UK), Andy Foote (NTNY, NO) , Michael C Fontaine
Collaborators: Marie Louis
learn more ...
Collaborators: Marie Louis
learn more ...
Details To be Announced
Project funding: University of Groningen, NL
[Photo credit: xxx]
Evolutionary genomics of Anopheles mosquitoes adaptation and speciation

PI : Michael C. Fontaine
Collaborators: Nora J. Besansky (U. Notre Dame, USA) ; Anopheles gambiae1000 genome consortium (Sanger, UK); François Sabot (DIADE, IRD); Anna-Sophie Fiston Lavier (ISEM, U. Montpellier); Jorge E Amaya-Romero; Clothilde Chenal.
learn more ...
Collaborators: Nora J. Besansky (U. Notre Dame, USA) ; Anopheles gambiae1000 genome consortium (Sanger, UK); François Sabot (DIADE, IRD); Anna-Sophie Fiston Lavier (ISEM, U. Montpellier); Jorge E Amaya-Romero; Clothilde Chenal.
learn more ...
Details To be Announced
Project funding: xxx
[Photo credit: xxx]
Evolutionary history of Plasmodium vivax in Americas (GENAD ANR JCJC, 2021-2024)
PI : Virginie Rougeron
Collaborators: Franck Prugnolle (CREES, MIVEGEC), Michael Fontaine (CREES, MIVEGEC), Josquin Daron (MIVEGEC), Céline Arnathau (CREES, MIVEGECE), Patrick Durand (CREES, MIVEGEC), Andrea Chaves (Universidad of Costa Rica, Costa Rica) and Camila Gonzalez Rosas (Universidad de Los Andes, Colombia). Students : Clarice Moulin (PhD student, 2020-2023) and Soledad Santillan (Master 2 student, 2020).
learn more ...
Collaborators: Franck Prugnolle (CREES, MIVEGEC), Michael Fontaine (CREES, MIVEGEC), Josquin Daron (MIVEGEC), Céline Arnathau (CREES, MIVEGECE), Patrick Durand (CREES, MIVEGEC), Andrea Chaves (Universidad of Costa Rica, Costa Rica) and Camila Gonzalez Rosas (Universidad de Los Andes, Colombia). Students : Clarice Moulin (PhD student, 2020-2023) and Soledad Santillan (Master 2 student, 2020).
learn more ...
Plasmodium vivax, the most prevalent human malaria parasite outside sub-Saharan Africa, represents an interesting model to study how pathogens adapt themselves to new environments. Indeed, the evolutionary history of this parasite is characterized by a succession of several colonization events on different primate species but also on different human populations. Very likely of Asian or African origin, P. vivax is indeed now present in almost all inter-tropical regions that it colonized more or less recently following human migrations. In the Americas, two events of colonization happened: (i) the first one when P. vivax arrived and infected new human populations and (ii) the second one with the transfer of P. vivax from humans to American monkeys, which gave rise to a new species genetically very close to P. vivax, named Plasmodium simium. During these colonisation events, P. vivax got exposed to new conditions in different vector species, in distinctive human populations and in different American monkey species. All these conditions likely exerted selective pressures on its genome to which it had to adapt by evolving towards new phenotypes and thus new genotypes.
One way to reconstruct the evolutionary history and predict genetic adaptation of parasites to new environments is to analyse whole genomes. Concerning P. vivax, although hundreds of strains from different parts of the world have been sequenced, genetic information about this parasite is still missing from several south American countries, mostly from the African continent (only few genomes have been published, with one from Mauritania, 4 from Madagascar and about 30 from Ethiopia) and from Eurasia (i.e. Turkey, Armenia, Azerbaidjan etc.). For P. simium, no genome has been yet published. Currently, the available genetic data are thus insufficient to robustly test evolutionary scenarios and study genetic adaptation of these parasites to new environments. Thus, one innovative aspect of this project will be to complete these last pieces of this “puzzle” by generating 372 genomes of P. vivax from missing countries in South America (N=69), Africa (N=189) and Eurasia (N=114) and to sequence the first P. simium genomes from other countries than Brazil (N=28).
GENERAL QUESTIONS. What is the evolutionary history of these parasites (P. vivax and P. simium) in the Americas? How, when and through which routes did they colonise the New World? How did they adapt to these new environments (new human populations, new vector species and new host species (Plathirrini monkeys))? Which genes have been involved?
Project funding: ANR
[Photo credit: xxx]
Evolutionary origin of Plasmodium vivax : a population genomic approach (PEPS ECOMOB MOV; 2020)
PI : Virginie Rougeron
Collaborators: Franck Prugnolle (CREES, MIVEGEC), Michael C. Fontaine (CREES, MIVEGEC), Josquin Daron (MIVEGEC), Clarice Mouline
learn more ...
Collaborators: Franck Prugnolle (CREES, MIVEGEC), Michael C. Fontaine (CREES, MIVEGEC), Josquin Daron (MIVEGEC), Clarice Mouline
learn more ...
Details To be Announced
Project funding: University of Groningen, NL
[Photo credit: xxx]
SOME RECENT PUBLICATIONS Click here for the full list
Global flyway evolution in red knots Calidris canutus and genetic evidence for a Nearctic refugium

Jesse Conklin, Yvonne Verkuil, Philip Battley, Chris Hassell, James Johnson, Job ten Horn, Pavel Tomkovich, Allan Baker, Theunis Piersma, Michael C. Fontaine
Molecular Ecology (in press, 2022)
click for abstract ...
Molecular Ecology (in press, 2022)
click for abstract ...
Present-day ecology and population structure are the legacies of past climate and habitat perturbations, and this is particularly true for species that are widely distributed at high latitudes. The red knot, Calidris canutus, is an arctic-breeding, long-distance migratory shorebird with six recognized subspecies defined by differences in morphology, migration behavior, and annual-cycle phenology, in a global distribution thought to have arisen just since the Last Glacial Maximum (LGM). We used nextRAD sequencing of 10,881 single-nucleotide polymorphisms (SNPs) to assess the neutral genetic structure and phylogeographic history of 172 red knots representing all known global breeding populations. Using population genetics approaches, including model-based scenario-testing in an approximate Bayesian computation (ABC) framework, we infer that red knots derive from two main lineages that diverged ca. 34,000 years ago, and thus persisted at the LGM in both Palearctic and Nearctic refugia, followed by at least two instances of secondary contact and admixture. In two flyways, we detected clear genetic structure between population pairs with similar migrations and substantial geographic overlap in the non-breeding season. Conversely, other populations were only weakly differentiated despite clearly divergent migratory phenotypes and little or no apparent contact throughout the annual cycle. In general, the magnitude of genetic differentiation did not match that of phenotypic differences among populations, suggesting that flyway-specific phenotypes developed quite rapidly and do not necessarily impose barriers to gene flow. Our results suggest that population structure and migratory phenotypes in red knots arose from a complex interplay among phylogeography, plasticity, and selective processes.
Population structure in a continuously distributed coastal marine species, the harbor porpoise, based on microhaplotypes derived from poor quality samples

Morin P. A., Forester B. R., Forney K. A., Crossman C. A., Hancock-Hanser B. L., Robertson K. M., Barrett-Lennard L. G., Baird R. W., Calambokidis J., Gearin P., Hanson B. M., Schumacher C., Harkins T., Fontaine M. C., Taylor B. L., Parsons K.
Molecular Ecology (accepted with minor revisions, 2021)
click for abstract ...
Molecular Ecology (accepted with minor revisions, 2021)
click for abstract ...
Harbor porpoise in the North Pacific are found in coastal waters from southern California to Japan, but population structure is poorly known outside of a few local areas. We used multiplexed amplicon sequencing of 292 loci and genotyped clusters of SNPs as microhaplotypes (N=271 samples) in addition to mtDNA sequence data (N=413 samples), to examine the genetic structure from samples collected along the Pacific coast and inland waterways from California to southern British Columbia. We confirmed an overall pattern of strong isolation-by-distance, suggesting that individual dispersal is restricted. We also found evidence of regions where genetic differences are larger than expected based on geographic distance alone, implying current or historical barriers to gene flow. In particular, the southernmost population in California is genetically distinct (FST = 0.02 (microhaplotypes); 0.31 (mtDNA)), with both reduced genetic variability and high frequency of an otherwise rare mtDNA haplotype. At the northern end of our study range, we found significant genetic differentiation of samples from the Strait of Georgia, previously identified as a potential biogeographic boundary or secondary contact zone between harbor porpoise populations. Association of microhaplotypes with remotely- sensed environmental variables indicated potential local adaptation, especially at the southern end of the species’ range. These results inform conservation and management for this nearshore species, illustrate the value of genomic methods for detecting patterns of genetic structure within a continuously distributed marine species, and highlight the power of microhaplotype genotyping for detecting genetic structure in harbor porpoises despite reliance on poor-quality samples.
Read moreNo leading-edge effect in North Atlantic harbor porpoises: Evolutionary and conservation implication
Yacine Ben Chehida, Roisin Loughnane, Julie Thumloup, Kristin Kaschner, Cristina Garilao, Patricia Rosel, Michael C. Fontaine
Evolutionary Applications (acceptedwith minor revisions, 2021)
BiorXiv preprint (2020), doi: https://doi.org/10.1101/2020.11.03.366542
click for abstract ...
Evolutionary Applications (acceptedwith minor revisions, 2021)
BiorXiv preprint (2020), doi: https://doi.org/10.1101/2020.11.03.366542
click for abstract ...
Understanding a species response to past environmental changes can help forecast how they will cope with ongoing climate changes. Harbor porpoises are widely distributed in the North Atlantic and were deeply impacted by the Pleistocene changes with the split of three sub-species. Despite major impacts of fisheries on natural populations, little is known about population connectivity and dispersal, how they reacted to the Pleistocene changes and how they will evolve in the future. Here, we used phylogenetics, population genetics, and predictive habitat modelling to investigate population structure and phylogeographic history of the North Atlantic porpoises. A total of 925 porpoises were characterized at 10 microsatellite loci and one-quarter of the mitogenome (mtDNA). A highly divergent mtDNA lineage was uncovered in one porpoise off Western Greenland, suggesting that a cryptic group may occur and could belong to a recently discovered mesopelagic ecotype off Greenland. Aside from it and the southern sub-species, spatial genetic variation showed that porpoises from both sides of the North Atlantic form a continuous system belonging to the same subspecies (Phocoena phocoena phoceona). Yet, we identified important departures from random mating and restricted intergenerational dispersal forming a highly significant isolation-by-distance (IBD) at both mtDNA and nuclear markers. A ten times stronger IBD at mtDNA compared to nuclear loci supported previous evidence of female philopatry. Together with the lack of spatial trends in genetic diversity, this IBD suggests that migration-drift equilibrium has been reached, erasing any genetic signal of a leading-edge effect that accompanied the predicted recolonization of the northern habitats freed from Pleistocene ice. These results illuminate the processes shaping porpoise population structure and provide a framework for designing conservation strategies and forecasting future population evolution.
Read moreRadiation with reticulation marks the origin of a major malaria vector
Small S. T., Labbé F., Lobo N. F., Koekemoer L. L., Sikaala C. H., Neafsey D. E., Hahn M. W., Fontaine M. C., Besansky N. J.
P. Natl. Acad. Sci. USA, 2020 vol. 26: 202018142
Doi: 10.1073/pnas.2018142117
click for abstract ...
P. Natl. Acad. Sci. USA, 2020 vol. 26: 202018142
Doi: 10.1073/pnas.2018142117
click for abstract ...
Advances in genomics have led to an appreciation that introgression is common, but its evolutionary consequences are poorly understood. In recent species radiations the sharing of genetic variation across porous species boundaries can facilitate adaptation to new environments and generate novel phenotypes, which may contribute to further diversification. Most Anopheles mosquito species that are of major importance as human malaria vectors have evolved within recent and rapid radiations of largely nonvector species. Here, we focus on one of the most medically important yet understudied anopheline radiations, the Afrotropical Anopheles funestus complex (AFC), to investigate the role of introgression in its diversification and the possible link between introgression and vector potential. The AFC comprises at least seven morphologically similar species, yet only An. funestus sensu stricto is a highly efficient malaria vector with a pan-African distribution. Based on de novo genome assemblies and additional whole-genome resequencing, we use phylogenomic and population genomic analyses to establish species relationships. We show that extensive interspecific gene flow involving multiple species pairs has shaped the evolutionary history of the AFC since its diversification. The most recent introgression event involved a massive and asymmetrical movement of genes from a distantly related AFC lineage into An. funestus, an event that predated and plausibly facilitated its subsequent dramatic geographic range expansion across most of tropical Africa. We propose that introgression may be a common mechanism facilitating adaptation to new environments and enhancing vectorial capacity in Anopheles mosquitoes.
Read moreSelection on ancestral genetic variation fuels parallel ecotype formation in bottlenose dolphins

Louis M., Galimberti M., Archer E., Berrow S., Brownlow A., Fallon R., Nykänen M., OBrian J., Robertson K.M., Rosel P.E., Simon-Bouhet B., Wegmann D., Fontaine M.C.*, Foote A.D.*, & Gaggiotti O.E.*
(*contributed equally as co-supervisors)
BiorXiv preprint (2020), doi: https://doi.org/10.1101/2020.10.05.325159v1
click for abstract ...
(*contributed equally as co-supervisors)
BiorXiv preprint (2020), doi: https://doi.org/10.1101/2020.10.05.325159v1
click for abstract ...
What are the mechanisms that allow species to extend their ranges and adapt to the novel environmental conditions they find in the newly available habitat? The study of parallel adaptation of pairs of populations to similar environments can provide great insights into this question. Here, we test for parallel evolution driven by niche specialization in a highly social marine mammal, the common bottlenose dolphin, Tursiops truncatus, and investigate the origins of the genetic variation driving local adaptation. Coastal ecotypes of common bottlenose dolphins have recurrently emerged in multiple regions of the world from pelagic ecotype populations, when novel habitat became available. Analyzing the whole genomes of 57 individuals using comparative population genomics approaches, we found that coastal ecotype evolution was relatively independent between the Atlantic and Pacific, but related between different regions within the Atlantic. We show that parallel adaptation to coastal habitat was facilitated by repeated selection on ancient alleles present as standing genetic variation in the pelagic populations. Genes under parallel adaptation to coastal habitats have roles in cognitive abilities and feeding. Therefore, parallel adaptation in long-lived social species may be driven by a combination of ecological opportunities, selection acting on ancient variants, and stable behavioural transmission of ecological specialisations. Tried and tested genetic variation that has been subject to repeated bouts of selection, may promote linked adaptive variants with minimal pleiotropic effects, thereby facilitating their persistence at low frequency in source populations and enabling parallel evolution.
Read moreGenome variation and population structure among 1142 mosquitoes of the African malaria vector species Anopheles gambiae and Anopheles coluzzii

The Anopheles gambiae 1000 Genomes Consortium
Genome Research (2020), doi: 10.1101/gr.262790.120
click for abstract ...
Genome Research (2020), doi: 10.1101/gr.262790.120
click for abstract ...
Mosquito control remains a central pillar of efforts to reduce malaria burden in sub-Saharan Africa. However, insecticide resistance is entrenched in malaria vector populations, and countries with a high malaria burden face a daunting challenge to sustain malaria control with a limited set of surveillance and intervention tools. Here we report on the second phase of a project to build an open resource of high-quality data on genome variation among natural populations of the major African malaria vector species Anopheles gambiae and Anopheles coluzzii. We analyzed whole genomes of 1142 individual mosquitoes sampled from the wild in 13 African countries, as well as a further 234 individuals comprising parents and progeny of 11 laboratory crosses. The data resource includes high-confidence single-nucleotide polymorphism (SNP) calls at 57 million variable sites, genome-wide copy number variation (CNV) calls, and haplotypes phased at biallelic SNPs. We use these data to analyze genetic population structure and characterize genetic diversity within and between populations. We illustrate the utility of these data by investigating species differences in isolation by distance, genetic variation within proposed gene drive target sequences, and patterns of resistance to pyrethroid insecticides. This data resource provides a foundation for developing new operational systems for molecular surveillance and for accelerating research and development of new vector control tools. It also provides a unique resource for the study of population genomics and evolutionary biology in eukaryotic species with high levels of genetic diversity under strong anthropogenic evolutionary pressures.
Read moreEurope as a bridgehead in the worldwide invasion history of grapevine downy mildew, Plasmopara viticola

Michael C. Fontaine, Frédéric Labbé, Yann Dussert, Laurent Delière, Sylvie Richart-Cervera, Tatiana Giraud, François Delmotte
Current Biology (In revision)
BiorXiv doi:10.1101/2020.09.22.307678
click for abstract ...
Current Biology (In revision)
BiorXiv doi:10.1101/2020.09.22.307678
click for abstract ...
Europe is the historical cradle of viticulture, but grapevines have been increasingly threatened by pathogens of American origin. The invasive oomycete Plasmopara viticola causes downy mildew, one of the most devastating grapevine diseases worldwide. Despite major economic consequences, its invasion history remains poorly understood. Comprehensive population genetic analyses of ~2000 samples from the most important wine-producing countries revealed very low genetic diversity in invasive downy mildew populations worldwide. All the populations originated from one of five native North American lineages, the one parasitizing wild summer grape. After an initial introduction into Europe, invasive European populations served as a secondary source of introduction into vineyards worldwide, including China, South Africa and, twice independently, Australia. Invasion of Argentina probably represents a tertiary introduction from Australia. Our findings provide a striking example of a global pathogen invasion resulting from secondary dispersal of a successful invasive population. It will help designing quarantine regulations and efficient breeding for resistance against grapevine downy mildew.
Read moreMitochondrial genomics reveals the evolutionary history of the porpoises (Phocoenidae) across the speciation continuum.
Yacine Ben Chehida, Julie Thumloup, Cassie Schumacher, Timothy Harkins, Alex Aguilar, Asunción Borrell, Marisa Ferreira, Lorenzo Rojas-Bracho, Kelly M. Robertson, Barbara L. Taylor, Gísli A. Víkingsson, Arthur Weyna, Jonathan Romiguier, Phillip A. Morin & Michael C. Fontaine
Scientific Reports, 10: 15190 (2020) doi:10.1038/s41598-020-71603-9
click for abstract ...
Scientific Reports, 10: 15190 (2020) doi:10.1038/s41598-020-71603-9
click for abstract ...
Historical variation in food resources is expected to be a major driver of cetacean evolution, especially for the smallest species like porpoises. Despite major conservation issues among porpoise species (e.g., vaquita and finless), their evolutionary history remains understudied. Here, we reconstructed their evolutionary history across the speciation continuum. Phylogenetic analyses of 63 mitochondrial genomes suggest that porpoises radiated during the deep environmental changes of the Pliocene. However, all intra-specific subdivisions were shaped during the Quaternary glaciations. We observed analogous evolutionary patterns in both hemispheres associated with convergent evolution to coastal versus oceanic environments. This suggests that similar mechanisms are driving species diversification in northern (harbor and Dall’s) and southern species (spectacled and Burmeister’s). In contrast to previous studies, spectacled and Burmeister’s porpoises shared a more recent common ancestor than with the vaquita that diverged from southern species during the Pliocene. The low genetic diversity observed in the vaquita carried signatures of a very low population size since the last 5,000 years. Cryptic lineages within Dall’s, spectacled and Pacific harbor porpoises suggest a richer evolutionary history than previously suspected. These results provide a new perspective on the mechanisms driving diversification in porpoises and an evolutionary framework for their conservation.
Read morePopulation genomic evidence of a Southeast Asian origin of Plasmodium vivax
J. Daron, A. Boissière, L. Boundenga, B. Ngoubangoye, S. Houze, C. Arnathau, C. Sidobre, J.-F. Trape, P. Durant, F. Renaud, M.C. Fontaine*, F. Prugnolle*, V. Rougeron* (*Co-supervisors of the study)
BiorXiv preprint (2020) doi:https://doi.org/10.1101/2020.04.29.067439
In revision in Science Advances
click for abstract ...
BiorXiv preprint (2020) doi:https://doi.org/10.1101/2020.04.29.067439
In revision in Science Advances
click for abstract ...
Plasmodium vivax is the most prevalent and widespread human malaria parasite, with almost three billion people living at risk of infection. With the discovery of its closest genetic relatives in African great apes (Plasmodium vivax-like), the origin of P. vivax has been proposed to be located in the sub-Saharan African area. However, the limited number of genetic markers and samples investigated questioned the robustness of this result. Here, we examined the population genomic variation of 447 human P. vivax strains and 19 ape P. vivax-like strains originating from 24 different countries across the world. We identified 2,005,455 high quality single-nucleotide polymorphism loci allowing us to conduct an extensive characterization to date of P. vivax worldwide genetic variation. Phylogenetic relationships between human and ape Plasmodium revealed that P. vivax is a sister clade of P. vivax-like, not included within the radiation of P. vivax-like. By investigating a variety of aspects of P. vivax variation, we identified several striking geographical patterns in summary statistics as function of increasing geographic distance from Southeast Asia, suggesting that P. vivax may derived from serial founder effects from a single origin located in Asia.
Read moreNEWS
[July 2020] At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum

Ut enim ad minima veniam, quis nostrum exercitationem ullam corporis suscipit laboriosam, nisi ut aliquid ex ea commodi consequatur? Quis autem vel eum iure reprehenderit qui in ea voluptate velit esse quam nihil molestiae consequatur, vel illum qui dolorem eum fugiat quo voluptas nulla pariatur.
more ...
MAIN COLLABORATORS

















PHOTO GALLERY

Habour porpoises
Photo: © XXX
Selection on ancestral genetic variation fuels parallel ecotype formation in bottlenose dolphins
Photo: ©Marie Louis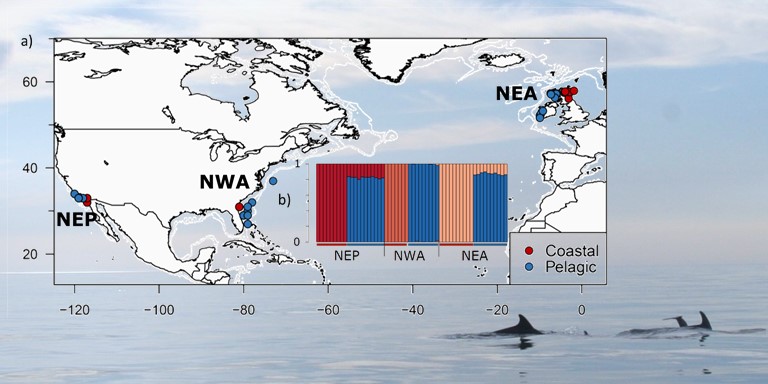
Selection on ancestral genetic variation fuels parallel ecotype formation in bottlenose dolphins
Photo: ©Marie Louis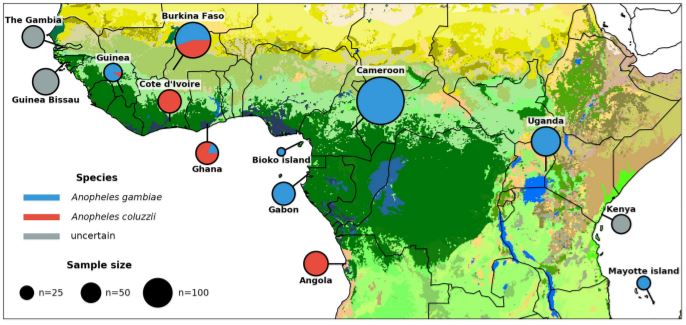
Genome variation and population structure among 1142 mosquitoes of the African malaria vector species Anopheles gambiae and Anopheles coluzzii
Photo: ©Ag1000G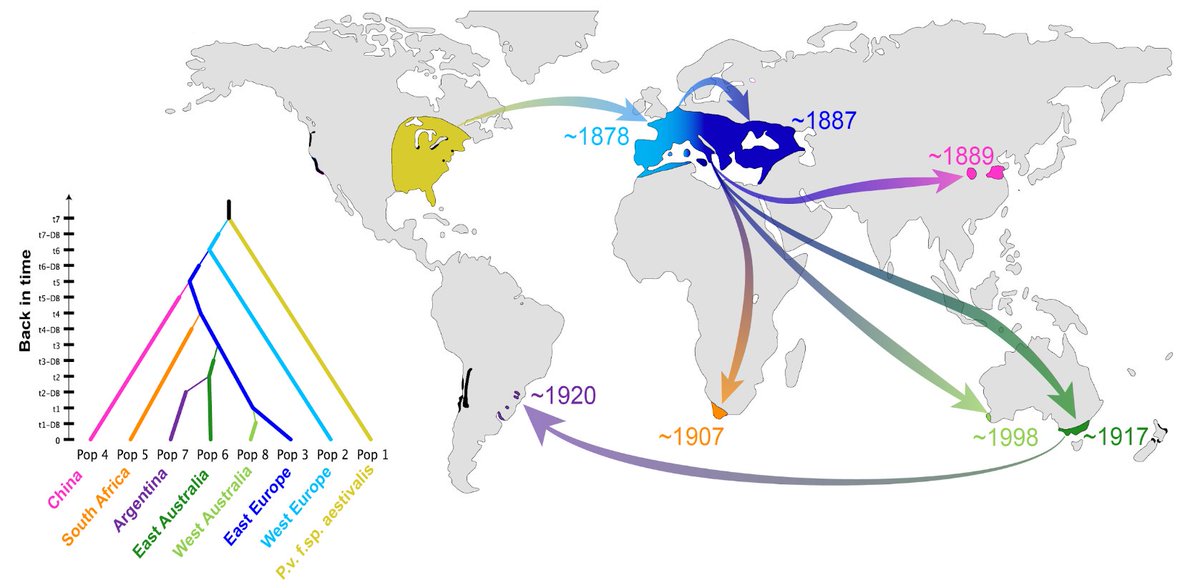
Europe as a bridgehead in the worldwide invasion history of grapevine downy mildew, Plasmopara viticola
Photo: ©Fontaine MC
TBA
Photo: © Centre for Research on the Ecology and Evolution of DiseaSes
Montpellier

Montpellier